
Diagnostic moléculaire
Pour soumettre des échantillons pour une ou plusieurs des analyses ci-dessous, veuillez utiliser le bon d'analyse CHUV-IMU no 151 (fourni sur demande au 021-3144107).
- Pour le diagnostic de la résistance et / ou du génotypage du virus de l'hépatite B (HBV), veillez à ce que l'ADN d'HBV soit détectable et complétez le champ des détails cliniques du bon d'analyse CHUV-IMU N ° 151.
- Des échantillons sanguins adéquats sont soit du sang EDTA-K, soit du plasma (5-10 ml, poste prioritaire, température ambiante). Le sérum a également été utilisé avec succès pour la détection de l'ARN des virus HDV et HEV.
- Le LCR peut être évalué dans les manifestations neurologiques présumées associées à l'infection par HEV. Dans ce cas, fournir toujours un échantillon apparié (plasma ou sérum) pour évaluer la synthèse intrathécale des anticorps.
Diagnostic moléculaire de la résistance d'HBV
Organisation génétique d'HBV
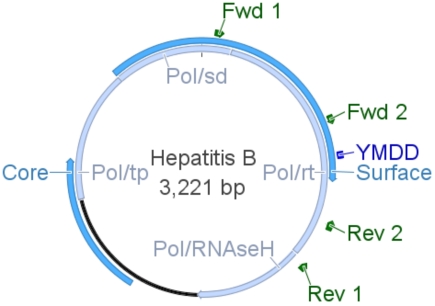
L'organisation génétique d'HBV est extrêmement compacte avec une partie du "core" et tous les gènes de surface (bleu) chevauchant le gène de la polymérase (bleu clair). Par conséquent, des mutations affectant un produit d'un gène peuvent affecter un autre gène. La polymérase est subdivisée en domaines fonctionnels: protéine terminale (tp), espaceur (sd), transcriptase inverse (rt) et RNAseH. Le site actif de la rt (YMDD, bleu foncé) et les séquences adjacentes sont la cible de mutations conférant une résistance aux médicaments antiviraux. La protéine "core" constitue la capside virale, tandis que l'antigène de surface constitue l'enveloppe du virus après ancrage dans une membrane lipidique d'origine cellulaire. D'autres caractéristiques importantes ne sont pas représentées ici par souci de simplicité: l'ARN pré-génomique utilisé comme matrice pour la replication de l'ADN viral, le cadre de lecture ouvert X dont la fonction est inconnue ainsi que les éléments régulateurs nécessaires à l'expression génique virale et à la replication de l'ADN viral. Les sites de liaison pour les amorces utilisées dans le diagnostic sont représentés en vert.
Identification moléculaire des mutations
L'identification des mutations ne peut être effectuée qu'après amplification par PCR de la rt à partir d'ADN extrait du plasma de patients infectés. Les amorces utilisées dans notre laboratoire sont représentées en vert sur le génome d'HBV présenté ci-dessus.
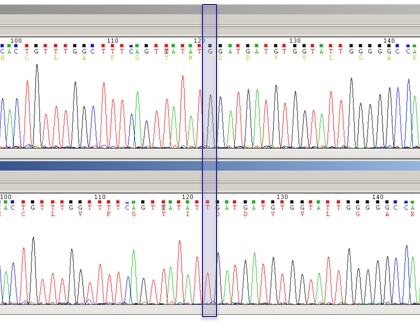
Une identification précise des mutations peut être effectuée par hybridation contre un panel de sondes (pour des mutations connues, sensibilité élevée de détection) ou par séquençage d'ADN (pour des mutations connues aussi bien que pour des mutations inconnues) à condition qu'elles représentent au moins 20% de la population des séquences. Ce niveau de détection est suffisant pour le diagnostic de routine. Une fois la séquence d'ADN déterminée, elle est traduite en acides aminés. Les mutations peuvent ensuite être identifiées après alignement de la séquence polypeptidique par rapport à des séquences de référence. Cette méthode, utilisée dans notre laboratoire, est illustrée ci-dessous à l'aide des chromatogrammes de séquençage de deux échantillons du même patient, avant (brin supérieur) et après échec de traitement avec la telbivudine (brin inférieur). La transversion G/T surlignée en bleu conduit à la mutation M204I connue pour conférer une résistance à ce médicament.
Quantification de l'ARN d'HDV
L'information génétique d'HDV est portée par une molécule circulaire d'ARN monocaténaire ayant une forte proportion d'appariements internes. Cette structure pose un défi aux techniques de PCR visant à l'évaluation quantitative de la virémie d'HDV dû à l'appariement intramoléculaire de nucléotides qui sont en compétition avec la liaison des amorces de la PCR ou de la transcription réverse. Pour surmonter cette difficulté, nous utilisons une PCR en temps réel après transcription inverse et des transitions rapides des étapes de la PCR. Ce test a été validé par rapport à des échantillons connus, et est très performant dans une étude Européenne de contrôle de qualité auquel nous avons participé récemment.
Quantification de l'ARN d'HEV
Nous avons adapté une PCR en temps réel à large spectre publiée par Jothikumar et. Al. (2006, J. Virol. Methods 131 (1): 65-71) pour surmonter la grande variabilité génétique d'HEV. Les amorces et la sonde sont régulièrement vérifiées vis-à-vis de la plupart des souches déposées dans GenBank, couvrant les 4 génotypes connus.
Le diagnostic d'HEV est compliqué par la courte durée de la virémie. Il est par conséquent préférable d'envoyer les échantillons le plus tôt possible après la manifestation des symptômes pour une sensibilité clinique maximale.


CHUV
Rue du Bugnon 48
CH-1011 Lausanne, Suisse
+41 21 314 4056

CHUV
Rue du Bugnon 48
CH-1011 Lausanne, Suisse
+41 21 314 4056