
Projects
The Riggi laboratory studies how the DNA in the human genome is packaged by a structure called chromatin. The genome consists of genes that code for the protein machines in our cells as well as regulatory elements that control those genes. The way a gene is organized into chromatin predicts whether it will be turned on or off, and thus make protein, in a particular cell type. A central question in human biology is how a single genome sequence can give rise to the hundreds of different cell types in the body. We now understand that differential patterns of gene expression underlie the many different cellular phenotypes seen in multicellular organisms. However, our understanding of how these gene expression patterns arise during development, how they are subsequently maintained in the adult organism and rewired to induce cancer development remains poor. A number of studies have indicated that these different expression patterns and phenotypes are intimately related to the way in which genomic DNA is organized into chromatin in the cell. This complex structure composed of proteins and DNA helps control which genes are expressed in a given cell type during a precise spatio-temporal pattern, and is critical to the function of normal cells. Moreover, a large body of evidence suggests that the “epigenome” is inappropriately altered in most, if not all, human cancers.
During the last decade the coordinated efforts of sequencing the genome of a comprehensive panel of primary human tumors has revealed fundamental differences between the biology of adult and pediatric tumors. Whereas adult tumors frequently display a significant number of genomic mutations, most of which are directly involved in the development of the tumor itself, pediatric tumors and a subcategory of human sarcomas are characterized by the presence of only few genetic alterations, believed to be the main drivers of these malignancies. Interestingly, the vast majority of the genes mutated in pediatric tumors and sarcomas are involved in chromatin organization, remodeling and cell fate transitions. For these reason, pediatric malignancies and sarcomas clearly represent an unique opportunity to investigate the role of epigenetic deregulation in cancer, in relative isolation from the large number of genetic mutations displayed by their adult counterparts. Therefore, the laboratory aim to investigate how single genetic events, driving pediatric tumors and sarcoma development, affect chromatin regulation to rewire cancer cells identities that contribute to cellular transformation.
The long-term goal of our research is the identification and characterization of pathways implicated in pediatric solid tumors and sarcomas. An important feature linking many of these malignancies is their strong association with developmental processes and, in particular, with the mechanisms that regulate the differentiation of stem cell populations during organogenesis. Our work combines tools in cell biology, biochemistry and molecular biology with next-generation sequencing to achieve increasingly precise, genome-wide views of chromatin structure, chromatin regulator binding and genome organization. Integrative analysis of chromatin state maps yields detailed annotations of the locations of functional elements in the human genome, including promoters, transcripts, silencers, insulators and enhancers. The group is particularly focused on understanding how dynamic alterations in chromatin structure and aberrant chromatin regulation contributes to cancer progression, heterogeneity and therapeutic resistance in pediatric solid tumors and sarcomas. In summary, we seek to identify common programs that would offer novel therapeutic options in these dismal diseases.
We have recently performed deep chromatin landscape profiling and analysis of primary Ewing sarcoma tumors, cell lines and precursor cells and have utilized this information to reconstruct functionally validated network models. Ewing sarcoma is the second most common bone malignancy in children and young adults, is a prototypical example of a pediatric tumor with a dominant genetic alteration in a transcriptional regulator. Ewing sarcoma is characterized in almost 90% of the cases by a chromosomal translocation that generates fusions between the EWS and FLI1 The EWS-FLI1 oncogenic fusion protein not only constitutes a defining diagnostic feature of Ewing sarcoma but also underlies its pathogenesis, and several studies have shown EWS-FLI1 to be crucial for the growth and survival of Ewing sarcoma cells and sufficient for the transformation of primary mesenchymal stem cells, a putative cell of origin. In our work we have found that EWS-FLI1 reprograms gene regulatory circuits in Ewing sarcoma by directly inducing or repressing enhancers. At GGAA repeat elements, which lack evolutionary conservation and regulatory potential in other cell types, EWS-FLI1 multimers induce chromatin opening and create de novo enhancers that physically interact with target promoters. Conversely, EWS-FLI1 inactivates conserved enhancers containing canonical ETS motifs by displacing wild-type ETS transcription factors. These divergent chromatin-remodeling patterns repress tumor suppressors and mesenchymal lineage regulators while activating oncogenes and potential therapeutic targets. Our findings demonstrate how EWS- FLI1 establishes an oncogenic regulatory program governing both tumor survival and differentiation.
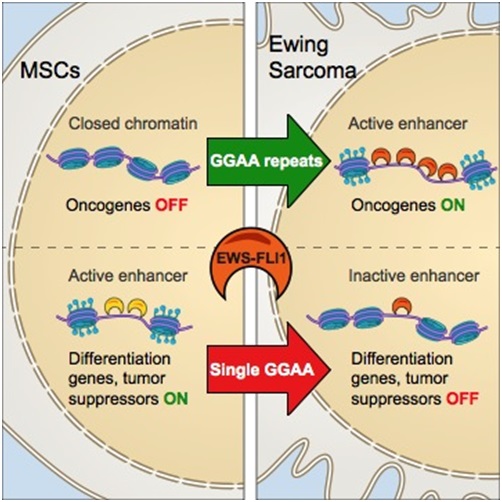
We are now testing the role of different target genes and collaborative chromatin regulators in the control of key survival pathways in Ewing sarcoma, and extending our epigenetic analysis to other pediatric and sarcoma tumors types, in order to identify and characterize their regulatory networks.


Rue du Bugnon 25
CH-1011 Lausanne, Vaud, Suisse
+41 21 314 7111

Rue du Bugnon 25
CH-1011 Lausanne, Vaud, Suisse
+41 21 314 7111