
Kuonen-LAB

Dr François Kuonen
MD-PhD, PD et MER1Médecin adjoint
+41 21 314 0377
Team
| Mathilde Chaffard | PhD student |
| Yiqun Zhang | PhD student |
| Sofia Tsiftsi | MD student |
| Victoria Prior | Master student |
William Stramke | Master student |
Major scientific contributions
The laboratory is interested in understanding the molecular determinants sustaining basal cell carcinoma (BCC) progression. During the last years, the laboratory identified:
The role of the microenvironment in BCC progression
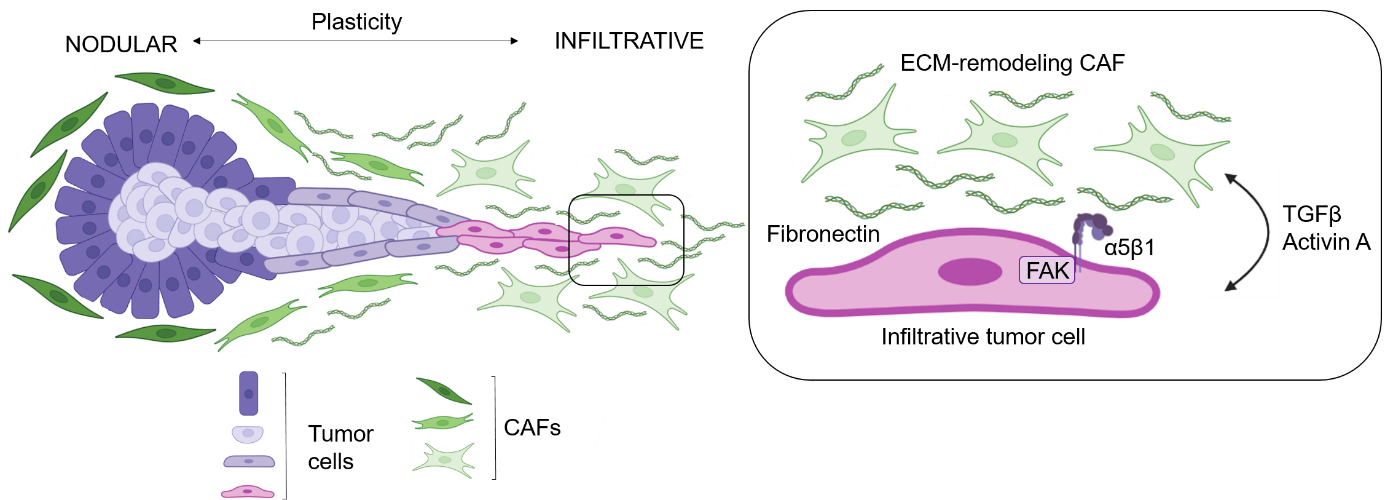
Based on their clinical and histological characteristics, thick BCC are typically divided into low-risk nodular and high-risk infiltrative subtypes, although the underlying mechanisms are poorly understood. We identified molecular mechanisms that explain the aggressiveness of high-risk infiltrative BCC, with a potential direct clinical impact. We showed that peritumoral TGFβ-induced fibronectin promotes adhesion and migration of BCC cells through integrin α5β1-mediated phosphorylation of focal adhesion kinase (FAK). Fittingly, we assembled functional compelling evidences for pre-clinical proof-of-principle of targeting tumor-CAFs interactions using FAK inhibitors to prevent BCC progression. Recently, we deciphered with unprecedented resolution the cellular and molecular governors of the invasive niche for progression, using a conceptual computational approach. In addition to TGFβ, we identified Activin A –mediated reciprocal transcriptional reprogramming in tumor cells and surrounding CAFs, providing novel potential therapeutic targets. Our work open important insights into the pathogenesis of aggressive infiltrative BCCs and identify integrin α5β1, focal adhesion kinase and Activin A signaling inhibition as promising strategies for the treatment of advanced BCCs.
Pathway switching as a mechanism of resistance to Hedgehog (HH) inhibitors
In 2012, inhibitors of the Hedgehog (HH) pathway were approved in the clinics for advanced BCCs, however with unexpectedly high rate of resistance. At this time, different groups around the world reported the striking arising of aggressive squamous cell carcinomas (SCCs) from BCCs previously treated with HH inhibitors. Using high-throughput sequencing, we identified resistant BCCs with a low HH pathway signature and concomitant Ras/MAPK pathway activation. This observation of reduced activation of the HH pathway in resistant BCCs contradicted previous demonstration that BCCs uniformly depend on the HH pathway for growth. Interestingly, driving constitutively active Ras in HH-responsive cell lines conferred resistance to HH pathway inhibitors, while conferring sensitivity to MAPK inhibitors. We since identified the HH to Ras/MAPK pathway switching in both spontaneous and HH inhibitors-driven BCC-to-SCC transition. In collaboration with Prof Oro at Stanford, we further interrogated the epigenetic/transcriptional plasticity underlying BCC-to-SC transition, and identified the prominent role of c-FOS-mediated transcriptional reprogramming, we could reverse using EGFR inhibitors. This seminal collaborative publication introduced the concept of the reversible, transcriptional basal-to-squamous transition, which prompted the reconsideration of the historical clear-cut distinction of BCCs and SCCs for a more fluidic relationship between keratinocyte-derived tumors.
Other fields of research
In addition, the laboratory is interested in clinical approaches to improve detection and therapy of skin tumors, with a particular focus on technologies improving oncological dermatologic surgery.
Selected Publications
More informations


CHUV
Avenue de Beaumont 29
CH - 1011 Lausanne, Vaud, Suisse
+41 21 314 1111

CHUV
Avenue de Beaumont 29
CH - 1011 Lausanne, Vaud, Suisse
+41 21 314 1111